
Analyse quality and quantity of DNA, RNA
Procedure
Simulator-Guided Step-by-Step Procedure of the Experiment
UV Spectrophotometer
A spectrophotometer is a laboratory instrument that measures the intensity of light passing through a solution and compares this to the amount of light entering the solution.
Preparing Samples for UV Absorbency Readings
Sample preparation for DNA concentration by UV spectrophotometry follow standard guidelines given in Current Protocols in Molecular Biology by Ausubel et. al. or Molecular Cloning: A Laboratory Manual by Sambrook, et al.
- Water should not be used to dilute the DNA because the pH must be precisely 7.4, and the pH of water can vary.
- TNE buffer is used for extremely accurate results.
- Temperature of the samples must be controlled because as temperature increases, the absorbance reading increases. Therefore, DNA samples and 1x TNE should be at room temperature.
- Only matching quartz cuvettes or cuvettes designed exclusively for UV spectrophotometry should be used as other types of cuvettes do not allow the UV wavelengths to pass through the sample correctly, giving an inaccurate reading.
- The cuvettes and solutions must be clean and free of particulates that can cause light scattering and give an inaccurate reading.
- To check-A reading of 1x TNE buffer only at 325 nm should give an absorbance of 0.01. If it is higher, the cuvette may be dirty or the TNE buffer may have particulates, requiring it to be made fresh and filtered.
Absorbance of UV light is measured at wavelength of 260 nm to determine double-stranded DNA concentration.
Fifty micrograms of double stranded DNA in one milliliter of buffer have an absorbance of 1.0 at this wavelength, but if there are other nucleic acids in the sample, the reading will be incorrect. This is because both single-stranded DNA and RNA absorb UV light at this wavelength.
- Meticulous technique in measuring the DNA is extremely important for accurate results. Use a small bore tip (the smallest tip available in the lab) and be sure to pre-rinse the tip with the DNA sample by pipetting it up and down a few times first.
- There should be no liquid on the outside of the tip, so wipe it with a Kim wipe before adding the DNA to the cuvette.
- The amount of buffer is determined by the design of the cuvette. With most quartz cuvettes, 2 ml of buffer must be added or the light path will be above, rather than through, the buffer. Add 1-2 μl of the DNA sample per ml of 1×TNE buffer and mix before inserting the clean cuvette into the spectrophotometer.
- Take several readings of each sample and average to calculate the concentration and be sure to use the dilution factor. For example, if the samples are diluted 1 μL into 1 mL three times and the absorbance readings were 0.0158, 0.0161, and 0.0180 the average is 0.0166 and the concentration of the DNA is 830 μg/ml (0.0166 x 50 ug/ml x 1000 d.f.).
- If the spectrophotometer has a program that does the calculation for you, take 3 readings and average the three calculated DNA concentrations. If readings fluctuate greatly from one reading to the next, the DNA concentration is probably too low to give accurate readings with the spectrophotometer you are using. Try adding more of the DNA sample to the cuvette (10 - 20 μL instead of 1 - 2 μL per mL buffer) and take the readings again.
The equation for calculation of DNA concentration: A260 x 50 μg/ml x dilution factor Nucleic Acid Purity.
- Protein in a sample can be detected at A280. The relative amount of protein contamination for a DNA sample can be determined by calculating the A260/A280 ratio.
- Ratios of 1.8 to 1.9 indicate that the DNA is highly purified. If the ratio is even slightly lower than 1.8, there is substantial protein contamination in the sample.
- Since both DNA and RNA absorb at A260, if there is RNA in the sample, the calculated concentration of double-stranded DNA will be inaccurately high.
- Running a sample on an agarose gel separates DNA from RNA (Figures 1 & 3) and can be used to determine if RNA is present. Contaminates such as EDTA (≥ 10 mM) and ethidium bromide can interfere with absorbance readings.
- The presence of protein, guanidine and other chaotropic salts or solvents, used in DNA and RNA purification methods for binding, absorb light at 230 nm and can lead to higher 260 nm absorbance measurements.
- This suggests that if contaminants are present and the A260 number is used for the calculation of yield, the amount of DNA may be overestimated.
- High quality DNA will have an A260/A280 ratio of 1.7-2.0.
- High quality RNA will have an A260/A280 ratio of ~2.0.
- DNA purity (protein contaminants) = A260 reading ÷ A280 reading
- Residual chaotropic salts and organic solvents, which can inhibit PCR, absorbs light in the 230 nm range. The A260/A230 ratio should be >1.5 for applications that are sensitive to chemical interference.
- The lower the ratio, the greater the number of organic compounds or chaotropic salts in the preparation.
DNA purity (chemical contaminants) = A260 reading ÷ A230 reading
Determination of DNA Quality and Quantity by Gel Electrophoresis
After the extraction procedure, the DNA is checked to verify that it is intact and clean of cellular contaminants. Determination of the quality and concentration can be accomplished in two ways: gel electrophoresis and UV spectrophotometry.
Gel Electrophoresis Method
- DNA can be diluted and run on an agarose gel. By running a molecular weight marker of known concentration, the extracted DNA concentration can be determined.
- The appearance of the DNA on the gel can also reveal if it is clean and intact.
- Agarose is a derivative of agar, a polysaccharide derived from algae. DNA fragments, including molecular weight markers, are most of the time heated to 65oC prior to electrophoresis to straighten any loops formed along the length of the molecules so that migration through the gel is uniform.
- In solution, ionization of the phosphate groups along the backbone of the DNA results in many negative charges on the molecule. Once loaded into the gel, an electric current is applied and the negatively charged molecules of DNA move through the gel toward the positive electrode.
- The gel is exposed to ethidium bromide, a flat molecule that intercalates, or slides between, the stacked base pairs of the DNA. Ethidium bromide fluoresces orange in UV light, making it possible to visualize the DNA
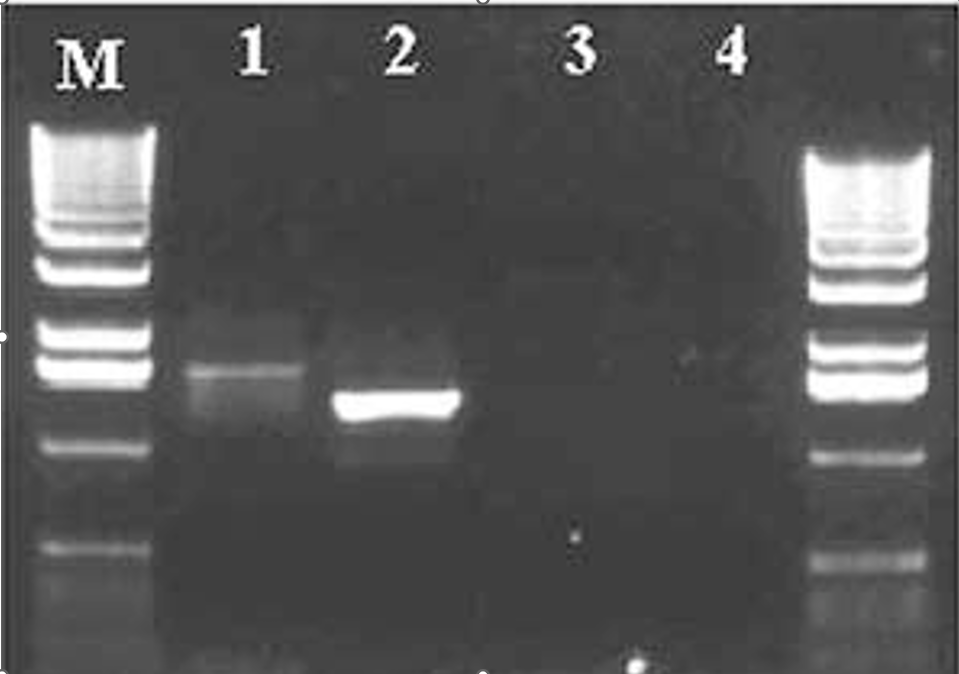 Figure 1. Bands of genomic DNA previously incubated with ethidium bromide fluoresce in UV light at 302 nm.
Figure 1. Bands of genomic DNA previously incubated with ethidium bromide fluoresce in UV light at 302 nm.
Lane M = Molecular weight marker DNA
The concentration of DNA can be estimated by running it on an agarose gel. It is best to dilute the DNA 1/10 and 1/100 and run both dilutions on the gel with a molecular weight marker (MWM) of known concentration.
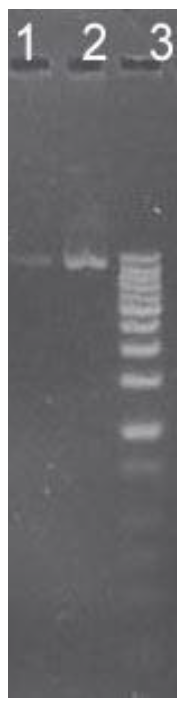 Figure 2. Calculation of DNA Concentration. 5 μl of a 1/100 dilution of DNA was loaded in Lane 1, 5 μl of a 1/10 dilution in Lane 2. The MWM band designated by the arrow contains 10 ng of DNA. What was the concentration in g/l of the sample of DNA before it was diluted?
Figure 2. Calculation of DNA Concentration. 5 μl of a 1/100 dilution of DNA was loaded in Lane 1, 5 μl of a 1/10 dilution in Lane 2. The MWM band designated by the arrow contains 10 ng of DNA. What was the concentration in g/l of the sample of DNA before it was diluted?
Answer: The band of DNA in Lane 1 is about the same intensity as the MWM band at the arrow, so it also contains 10 ng. Since this is a 1/100 dilution, the original sample contained 100x as much. Since 5 μl were loaded on the gel, the concentration of the original sample is: 1 g/5 μl = 0.2 g/μl.
The appearance of the DNA on the gel can also reveal if it is clean and intact. If the DNA is intact, it will appear as a distinct band on the gel. If it is degraded, it will appear as a smear of thousands of small fragments. If the DNA is contaminated with protein, there will be a bright band of DNA at the bottom of the well and along the migration path from the wells where the slower moving protein trapped DNA. If the well is over loaded with DNA that is too concentrated, the band will have a jagged smear above it. See Figure 3.

Figure 3(a) and 3(b). Lane 1, molecular weight marker DNA; Lane 2, degraded DNA; Lane 3, intact chromosomal DNA; Lane 4, overloaded well.
Below, Lanes 4-6, DNA is contaminated with protein, some DNA is degraded, and overloading is evident in lanes 2-3. RNA is seen at arrow.
Ethidium bromide is a strong mutagen. Gloves must always be worn when handling gels or buffers containing this chemical.
Boiling agarose can cause burns. Wear hot gloves when removing agarose from hot plate or microwave oven.
The electric current in a gel electrophoresis chamber is extremely dangerous. Never remove a lid or touch the buffer once the power is turned on. Make sure the counter where the gel is being run is dry.
UV light, used to illuminate the DNA stained with ethidium bromide, is dangerous. Eye protection must be worn.